分享一篇近期发表在 Genome Biology 上 的一个基因家族分析软件:Seqrutinator。该软件用于识别和去除基因家族数据集中的无功能基因,包括假基因、测序错误、基因结构错误、比对错误等,从而避免基因家族鉴定中的假阳性结果,进一步确保基因家族注释的准确性,以便于后续系统发育分析和功能聚类。
背景意义
目前,基因家族鉴定流程并无唯一标准,但大多是基于 Blast 和 Hmmer,再辅以手工校正。无标准鉴定流程,导致不同流程、不同参数、不同校正过滤标准,都会对同一基因家族鉴定数目和基因上存在差异,从而对后续结果产生影响。
准确构建超家族系统发育树和进行多序列比对(MSAs)仍然是一个主要研究领域。为了提高MSA的质量,研究人员经常修剪掉不太可靠的列,但这可能会丢失信息。此外,非功能性同源物(Non-Functional Homologues, NFHs)提供的序列信息通常被视为噪声,可能会干扰MSA的正确处理。
Seqrutinator的设计
Seqrutinator的设计理念是提供一个目标序列审查方法,用于检测和移除NFHs。该工具通过四个假设来识别NFHs:短序列不太可能是功能性的;NFHs可能在MSA中引起大量连续的间隙丰富的列;NFHs可能在MSA中显示出大量连续的间隙;以及与超家族的HMMER剖面相比,伪基因和其他类似序列可能具有低相似性和低分数。
Seqrutinator由五个模块组成,包括短序列移除器(SSR)、非同源命中移除器(NHHR)、间隙引发器移除器(GIR)、连续间隙序列移除器(CGSR)和伪基因移除器(PR)。用户可以选择模块、它们实施的顺序以及影响自动化审查和过滤严格性的设置。
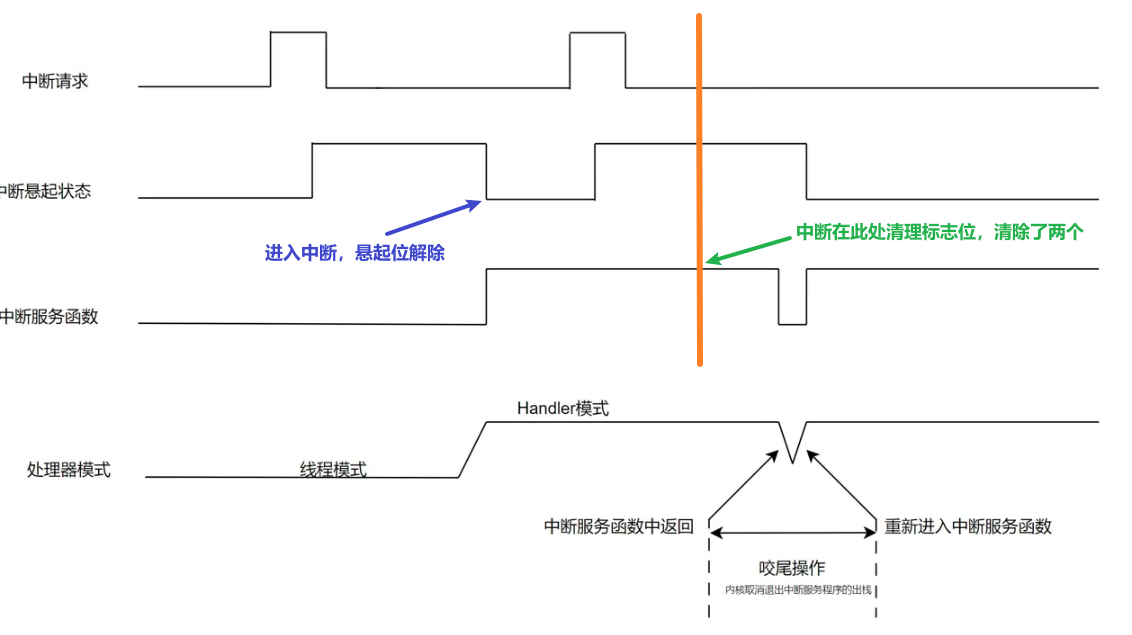
Seqrutinator 流程示意图
Seqrutinator的性能评估
研究者使用了19个完整的蛋白质组数据集,包括16种植物物种、两个版本的拟南芥蛋白质组以及两个版本的SwissProt数据集。通过Seqrutinator处理后,发现在拟南芥蛋白质组中移除的序列数量相对较少,而在最近发布的松树蛋白质组中移除的序列数量超过80%。这表明Seqrutinator能够一致地识别和移除NFHs。
Seqrutinator的应用效果
Seqrutinator处理后,MSA的质量得到了显著提高。通过使用BMGE工具修剪MSA,发现处理后的MSA中可靠列的数量显著增加。此外,Seqrutinator处理后的序列集在进行HMMERCTTER聚类分析时,产生了更大和更少的聚类,这表明Seqrutinator有效地去除了NFHs,而不是功能性同源物(Functional Homologues, FHs)。
讨论与结论
Seqrutinator是一个有效的工具,可以帮助自动化地挖掘蛋白质超家族序列,并且只要序列是真正同源的,它就能提供良好的结果。对于更复杂的情况,应该将序列分割成真正同源的子集以获得最佳结果。Seqrutinator的开发和测试表明,它是一个灵活且一致的工具,能够在不同数据集上识别和移除NFHs,从而改善MSA的质量,并为后续的系统发育分析和功能聚类提供更准确的数据集。
数据和材料的可用性
所有数据集均来自公共序列集。Seqrutinator软件(包括辅助脚本MuFasA和SeqYNet)代码可以在GitHub或Zenodo的存储库中免费下载。
Github:https://github.com/BBCMdP/Seqrutinator